Designing an Experiment
Designing a validation study for any kind of tool can be a great challenge. For cutting edge technology, this challenge can be even greater. In this blog post I’ll try to cover some problematic aspects and methodological challenges that one can bump into when validating an NGS based HLA typing method.
We don’t even have fool’s gold
Similarly to sequencing in general, Sanger sequencing based methods are still considered as a gold standard for HLA genotyping. Although this is not without a reason, generating high resolution HLA types with high accuracy using solely Sanger sequencing gets harder and harder as the number of known HLA alleles increases. There are also some general technical difficulties, caused simply by the nature of the method. For example, phasing between heterozygous positions can be difficult or even impossible. Due to the length limitations of Sanger sequencing, usually only a subset of the gene regions is covered even when multiple amplicons are sequenced, this can lead to additional difficulties during allele selection. People make mistakes, samples get mixed up, the DNA doesn’t always end up in the correct tube, random errors can occur. So, to avoid catastrophe (and also random positive effects), it is always wise to have redundancy. I.e. sequence and genotype at least a subset of your samples twice (or even more times) and make sure to have repeats when needed (e.g. within and between manufacturing lots). It also makes sense to make technical repeats as independent as possible, otherwise you might end up measuring how good your lab tech is. Although, in general, it might seem like a good idea to do validation on random samples, there are some aspects that must be considered when validating HLA data: 2. Allele frequencies of a single allele can vary greatly between different populations. So it might happen that a method works great in one population, but fails miserably in another. 3. Common alleles can also greatly vary between populations. 4. Don’t forget the special little snowflakes (e.g. rare alleles, alternatively expressed alleles, homozygous loci). 5. Chicken or egg problem: high resolution typing for all loci would be needed for sample selection, but high resolution typing is not available before going through the whole workflow. – By Krisztina Rigó (to be continued…)
Anything that can go wrong, will go wrong
Diversify your bonds
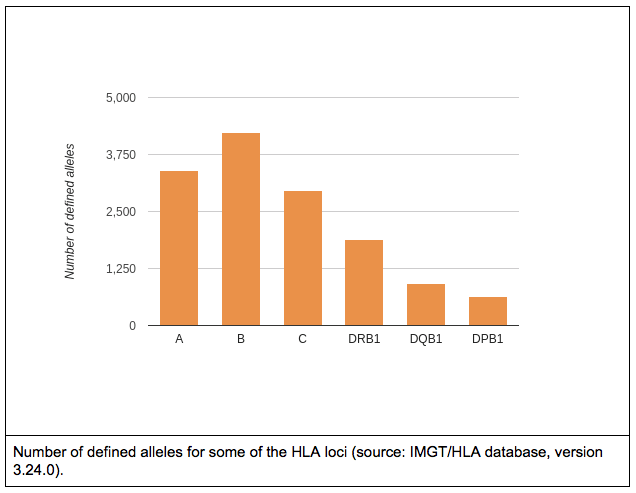
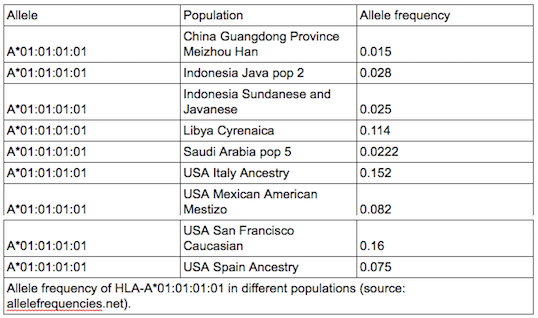

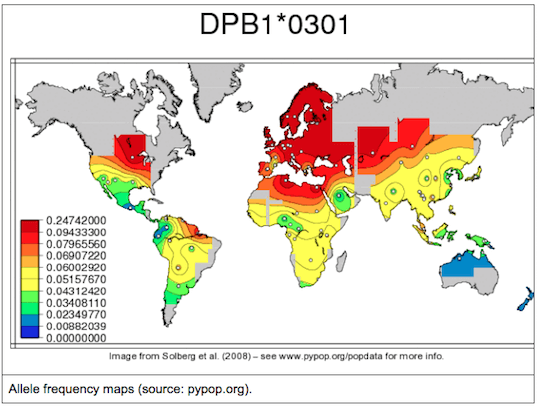


In an ideal world…
In reality…
So what should we do?




